Sara Duque González1, Carmen Luz Marrero Pérez1, Orlando Mesa Medina1, Alicia Herminia Callejón Callejón1, Antonio López Figueroa2.
Servicios de 1 Pediatría y de 2 Radiología. Complejo Hospitalario Universitario Nuestra Señora de Candelaria, Santa Cruz de Tenerife
Resumen
La malformación congénita de la vía aérea pulmonar, anteriormente conocida como malformación adenomatoidea quística pulmonar, es una entidad poco frecuente que se caracteriza por la formación durante la embriogénesis de quistes en diferente número y tamaño que sustituyen al parénquima pulmonar normal1. Se calcula una incidencia anual de 56 casos por cada 100.000 recién nacidos vivos2.
Aunque la mayor parte de los casos cursan de forma asintomática al nacimiento, puede verse comprometida la vía aérea. De ahí, la importancia del diagnóstico precoz mediante ecografía prenatal para prever la evolución y planificar el tipo de parto y la atención posterior del recién nacido.
En el presente artículo, se realiza una revisión bibliográfica de esta malformación pulmonar a raíz de un caso diagnosticado de forma prenatal en nuestro hospital. Se trata de un recién nacido pretérmino de 30 semanas en el que se objetivó en la ecografía selectiva una imagen compatible con malformación de la vía aérea pulmonar tipo 1 en el pulmón derecho.
Palabras clave: malformación congénita de la vía aérea pulmonar, quistes pulmonares
Congenital pulmonary airway malformation. Report of clinical case
Summary
Congenital pulmonary airway malformation, previously known as pulmonary cystic adenomatoid malformation, is a rare episode characterized by the formation of cysts of varying size and distribution during embryogenesis that replace normal pulmonary parenchyma1. An annual incidence of 56 cases per 100,000 live newborns is calculated2.
Although most cases are asymptomatic at childbirth, the airway might get involved, hence the importance of early diagnosis through prenatal ultrasound as to anticipate the evolution and plan the of child birth and subsequent care of the newborn can be organized.
In this paper, a bibliographic review of this pulmonary malformation is carried out as a result of an early diagnosed case in our hospital. This is a 30 week preterm newborn in which an image consistent with type 1 pulmonary airway malformation in the right lung was observed on selective ultrasound.
Key words: congenital pulmonary airway malformation congenital, lung cysts
Introducción
Las malformaciones congénitas pulmonares son un grupo heterogéneo de enfermedades que se caracterizan por la obstrucción de la vía aérea intraútero, lo que ocasiona el desarrollo anormal del árbol traqueo-bronquial. Tanto la definición como la clasificación de las diferentes entidades resulta compleja, ya que en muchas ocasiones se solapan desde el punto de vista clínico, radiológico e histológico1,2.
Presentamos una caso de un recién nacido pretérmino de 30 semanas en el que se objetivó en la ecografía selectiva una imagen compatible con malformación de la vía aérea pulmonar tipo 1 en el pulmón derecho.
Caso clínico
Presentamos el caso de un recién nacido pretérmino de 30 semanas con un diagnóstico prenatal de malformación congénita de la vía aérea pulmonar derecha de tipo 1.
Se trata de la tercera gestación de una madre de 38 años con asma, psoriasis, neuralgia del trigémino e hipotiroidismo subclínico como antecedentes pregestacionales relevantes. Como hallazgos de interés, el cribado del primer trimestre mostró un alto riesgo de trisomía del par 21 y de preeclampsia por el que se indicó tratamiento con ácido acetilsalicílico.
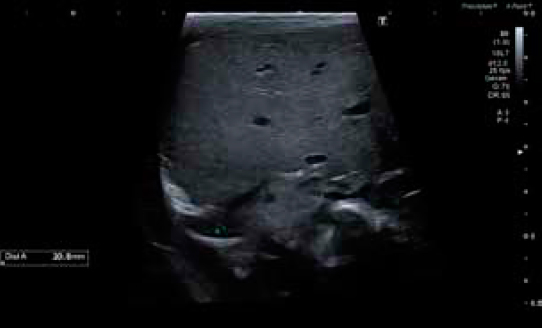
En la ecografía selectiva, se objetivó una malformación congénita de la vía aérea pulmonar tipo 1 (quiste único) en la base derecha. Se realizó una amniocentesis dada la asociación con cromosomopatías, con estudio genético y cariotipo normales. Se realizaron ecografías seriadas en las que se objetivó, inicialmente, un incremento de tamaño del quiste, para disminuir en controles posteriores hasta la semana 26+3 donde se realizó la última ecografía preparto en la que se observó un volumen de 30×17,8×13,5 mm, sin signos de descompensación hemodinámica.
Su madre requirió ingreso en dos ocasiones por amenaza de parto pretérmino, por lo que se indujo una maduración pulmonar completa y se administró una dosis más de corticoide seis días antes del parto. En la semana 30, comenzó nuevamente la dinámica uterina y se realizó una cesárea electiva por presentación podálica.
El neonato nació sin esfuerzo respiratorio, hipotónico y con frecuencia cardíaca entre 60-100 lpm. Se inició ventilación con doble presión. Ante la ausencia de respuesta se procedió a una intubación orotraqueal según protocolo de reanimación cardiopulmonar avanzada. Una vez estabilizado, se trasladó a la Unidad de Cuidados Intensivos Neonatales sin incidencias (pH umbilical 7,22, prueba de Apgar 3/6/7).
Recibió una dosis de surfactante alveolar a la hora de vida, con buena respuesta, lográndose disminuir el aporte de oxígeno. A las 24 horas de vida se extubó con éxito, permaneciendo con ventilación mecánica no invasiva durante 26 horas. Hasta los 23 días de vida (33+2 semanas de edad post-menstrual (EPM), precisó oxigenoterapia de forma intermitente para mantener unas saturaciones adecuadas.
Durante su estancia en la Unidad se realizó una radiografía de tórax al ingreso (figura 1) y controles ecográficos seriados (figuras 2 y 3) en los que se objetivó la lesión quística en la base del pulmón derecho. Se mantuvo una actitud expectante dada la estabilidad clínica del paciente.
De forma paralela, se diagnosticó al nacimiento una malformación asociada de la porción cartilaginosa de las costillas flotantes del hemitórax izquierdo consistente en una longitud mayor de lo habitual e inserción a un nivel superior en el esternón. Fue valorado por la Unidad de Rehabilitación que inició fisioterapia respiratoria de forma precoz y realizó vendajes funcionales para optimizar el movimiento de ambos hemidiafragmas, siendo una medida eficaz en la maduración del patrón respiratorio.
Como única complicación respiratoria asociada a la prematuridad, el paciente presentó apneas centrales que requirieron tratamiento con citrato de cafeína hasta las 34 semanas de EPM.
Finalmente, se procedió al alta hospitalaria a los 35 días de vida (EPM de 35 semanas) a la espera de ampliar el estudio de imagen con una TC para decidir la actitud terapéutica.
Discusión
Las principales malformaciones pulmonares son el secuestro pulmonar, el enfisema lobar congénito, el quiste broncogénico y la malformación congénita de la vía aérea pulmonar (también conocida como malformación adenomatoidea quística pulmonar). Esta última entidad es lamás frecuente3 y la que nos ocupa en el presente artículo. A pesar de ello, su incidencia es baja y se estima en 56 casos por cada 100.000 recién nacidos vivos2.
En relación a la patogenia, se cree que es el resultado de una desproporción entre la proliferación celular y la apoptosis, dando lugar a la formación de quistes en diferente número y tamaño2. Según la localización de los mismos y las características histológicas, pueden diferenciarse cinco subtipos de acuerdo con la clasificación de Stocker 20024. La tipo 1 la más frecuente y se caracteriza por un quiste único de gran tamaño (hasta 10 cm) que se origina a nivel del bronquio o del bronquiolo proximal o por múltiples quistes mayores de 2 cm de diámetro.
En la etapa prenatal, los quistes pueden comprimir los órganos adyacentes y comprometer el desarrollo adecuado de los mismos, produciendo polihidramnios al comprimir el esófago, limitación del crecimiento gástrico, hipoplasia pulmonar y, en casos más graves, hidrops fetalis por compresión de la vena cava1, entre otras manifestaciones.
Al nacimiento, la mayor parte de los casos son asintomáticos. Solo un 25 % presentan síntomas, por lo general en forma de dificultad respiratoria, aunque algunos debutan con un neumotórax espontáneo por rotura de los quistes. Desde que las mejoras técnicas han permitido diagnosticar un mayor número en el periodo prenatal, dicho porcentaje de casos sintomáticos ha disminuido5-6. El resto puede pasar desapercibido hasta la edad adulta y diagnosticarse como un hallazgo incidental al realizarse una prueba de imagen por otro motivo.
Actualmente, los avances técnicos en la ecografía prenatal han permitido que pueda establecerse la sospecha diagnóstica intraútero, generalmente a partir de la semana 16-202. En base a una serie de mediciones, puede preverse el curso de la malformación y establecerse el pronóstico7. En aquellos casos en los que existan dudas en el diagnóstico, puede ser necesario realizar otras pruebas complementarias, siendo la resonancia magnética nuclear (RMN) la más utilizada8.
Una vez establecida la sospecha diagnóstica intraútero, se requiere un seguimiento estrecho realizando controles ecográficos seriados con una periodicidad semanal, o diaria en caso de estadios iniciales de hidrops. En casos de una alta sospecha de cromosomopatía, estará indicada también la realización del estudio genético ya que servirá para establecer el pronóstico y la planificación del consejo genético. Sin embargo, no se realiza de forma rutinaria9.
Tras el parto, se realizará una radiografía de tórax, incluso en aquellos casos en lo que se haya descrito la resolución del cuadro por ecografía prenatal y se recomienda ampliar el estudio de imagen mediante tomografía computarizada (TC) o RMN, sobre todo de cara al tratamiento quirúrgico, siendo útil en ocasiones el uso de contraste para el diagnóstico diferencial. En pacientes asintomáticos, el estudio puede realizarse de forma diferida entre los 3-6 meses, pero en casos sintomáticos o con alto riesgo de complicaciones, deberá realizarse de forma inmediata10.
En aquellos pacientes que requieren tratamiento intraútero, no existe una indicación clara del momento óptimo para realizarlo sino que es una decisión que se basa en aspectos clínicos, de imagen, edad gestacional y cariotipo fetal8. Los pacientes que desarrollan hidrops fetalis pueden requerir tratamiento corticoideo intraútero e incluso, en casos de mala o nula respuesta, puede ser necesario el drenaje quirúrgico, la resección de los quistes o la inducción del parto.
Al nacimiento, en pacientes asintomáticos puede mantenerse una actitud expectante, observándose en muchos casos la resolución de los quistes de forma espontánea. Los pacientes sintomáticos, sin embargo, será sometidos a cirugía de forma precoz para evitar complicaciones futuras.
A pesar de que la mayor parte de los casos de malformación de la vía aérea pulmonar no presentan clínica al nacimiento, es necesario el diagnóstico prenatal para considerar las complicaciones que puedan derivar de la misma. Esto ayuda, entre otros aspectos, a gestionar los recursos necesarios para la estabilización inicial y manejo posterior del neonato, ya que el parto debería producirse en un hospital de tercer nivel con una Unidad de Cuidados Intensivos Neonatales y un Servicio de Cirugía Pediátrica especializado en el manejo de los casos graves de esta patología6.
Resulta fundamental realizar un manejo multidisciplinar, implicando a ginecólogos que realicen el seguimiento durante el embarazo, neonatólogos y cirujanos pediátricos que se encarguen del manejo inmediato tras el parto; radiólogos experimentados para valorar las pruebas de imagen que se soliciten; y neumólogos pediátricos que realicen el seguimiento al alta hospitalaria.
En aquellos neonatos asintomáticos en los que se decide el seguimiento periódico de la malformación, se han descrito como complicaciones la probabilidad de desarrollar infecciones pulmonares recurrentes y la capacidad de malignización (sobre todo, en la tipo 4) con el inconveniente de la exposición a la radiación dadas las pruebas de imagen de control.
Sin embargo, se han descrito remisiones espontáneas que podrían justificar el beneficio de mantener una actitud expectante, aunque dichos casos no están bien documentados11. En los pacientes sintomáticos, por el contrario, la indicación de cirugía es clara y se realiza de forma inmediata.
Actualmente, no existe una recomendación estándar sobre el manejo de estos pacientes, sino que la actitud dependerá de la estabilidad clínica y de las características de la lesión.
Bibliografía
- San Vicente B, Bardají C, Obiols P, Abad P, Rigol S. Malformación adenomatoidea quística: ¿somos capaces de prever su evolución? Cir Pediatr 2009; 22:87-92
- Mondéjar López P, Sirvent Gómez J. Malformaciones pulmonares congénitas. Malacia y otras malformaciones congénitas de la vía aérea. Protoc diagn ter pediatr 2017; 1:273-297
- Giubergia V. Malformaciones pulmonares congénitas. Neumol Pediatr 2014; 9:88-94
- Stocker JT. Congenital pulmonary airway malformation: A new name for and an expanded classification of congenital cystic adenomatoid malformation of the lung. Histopathology 2002; 41 (Supl. 2):424-430
- Parikh DH, Rasiah SV. Congenital lung lesions: Postnatal management and outcome. Semin Pediatr Surg 2015; 24:160-167
- Adzick NS, Harrison MR, Crombleholme TM, Flake AW, Howell LJ. Fetal lung lesions: Management and outcome. Am J Obstet Gynecol 1998; 179:884-889
- Crombleholme TM, Coleman B, Hedrick H, Liechty K, Howell L, Flake AW et al. Cystic adenomatoid malformation volume ratio predicts outcome in prenatally diagnosed cystic adenomatoid malformation of the lung. J Pediatr Surg 2002; 37:331-338
- Di Prima FAF, Bellia A, Inclimona G, Grasso F, Teresa M, Cassaro MN. Antenatally diagnosed congenital cystic adenomatoid malformations (CCAM): research review. J Prenat Med 2012; 6:22-30
- Hellmund A, Berg C, Geipel A, Bludau M, Heydweiller A, Bachour H et at. Prenatal diagnosis and evaluation of sonographic predictors for intervention and adverse outcome in congenital pulmonary airway malformation. PloS ONE. 2016; 11: e0150474.
- Sauvat F, Michel JL, Benachi A, Emond S, Revillon Y. Management of asymptomatic neonatal cystic adenomatoid malformations. Eur J Pediatr Surg 2003; 38:548-552.
- Cavoretto P, Molina F, Poggi S, Davenport M, Nicolaides KH. Prenatal diagnosis and outcome of echogenic fetal lung lesions. Ultrasound Obstet Gynecol 2008; 32:769-783
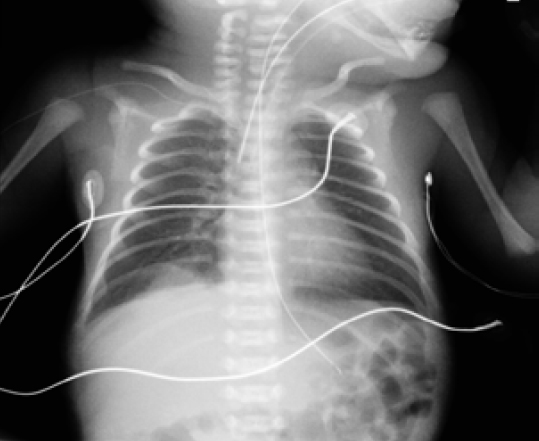
Figura 1. Radiografía torácica realizada al nacimiento en la que se objetiva la imagen de la consolidación a nivel de la base pulmonar derecha (lugar de localización de la malformación) Además, se objetiva la anomalía costal en la base pulmonar izquierda, con unos cartílagos costales con mayor longitud de lo habitual.
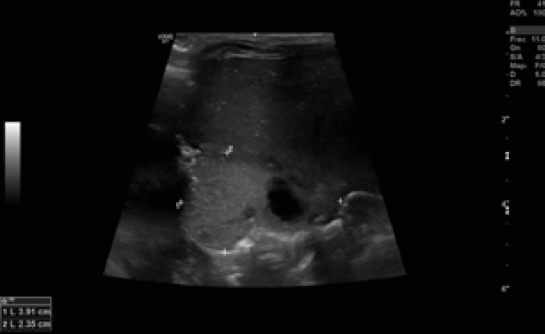
Figura 2. Imagen ecográfica en la que se objetiva una consolidación del parénquima pulmonar, sin signos de ventilación, con unas dimensiones aproximadas de 3,9 x 2,3 x 3 cm y cuatro quistes en su interior, el mayor bilobulado de 15 x 18 mm y el resto de 5 mm.