Eduardo Valerio Hernández1, Alejandra González Delgado2, Carmen Rosa Rodríguez Fernández-Oliva3, Mercedes Murray Hurtado4, Mónica Ruiz Pons Mónica5, Luis Peña Quintana6
1Pediatra de Atención Primaria en el Centro de Salud Casco-Botánico (Puerto de la Cruz). Gerencia de Atención Primaria de Santa Cruz de Tenerife. 2Bioquímica clínica. Responsable del laboratorio de cribado neonatal de Canarias, HUC. 3Pediatra. Coordinadora Regional de Pediatría Hospitalaria y de Atención Primaria. 4Pediatra. Unidad de Nutrición y Metabolopatías del Hospital Universitario de Canarias. 5Pediatra. Unidad de Nutrición y Matabolopatías del Hospital Universitario Nuestra Señora de Candelaria. 6Pediatra. Unidad de Gastroenterología y Nutrición Pediátrica. Hospital Universitario Materno-Infantil de Las Palmas. CIBEROBN ISCIII. ACIP. Universidad de Las Palmas de Gran Canaria
Resumen
El cribado neonatal ampliado (CNA) en Canarias avanza hacia la detección masiva a nivel bioquímico de metabolopatías por Espectrometría de tándem en masas (MS/MS). En el año 2022 se incluyeron la Enfermedad de la orina con olor a jarabe de arce (MSUD) y la homocistinuria (HCY) y en este 2023, se incluirá el cribado de la tirosinemia tipos 1 y 2. Importantes enfermedades con pronósticos totalmente diferentes si son cribadas y detectadas a tiempo, y cuyos tratamientos iniciados de forma temprana, además de alguno específico como es el caso de la tirosinemia tipo 1, son, como en todas las metabolopatías, esenciales para el adecuado control clínico y bioquímico de los pacientes.
Palabras clave: metabolopatías, cribado neonatal ampliado, enfermedad de la orina con olor a jarabe de arce, homocistinuria, tirosinemia, nitisinona
Expanded newborn screening in Canary Islands (2nd part). Maple syrup urine disease, Homocystinuria and Tyrosinemia type 1 and 2
Abstract
Expanded newborn screening in Canary Islands is moving towards mass biochemical detection of metabolopathies using tandem mass spectrometry (MS/MS). In 2022, Maple Syrup Urine Disease (MSUD) and homocystinuria (HCY) were included, and in 2023, screening for tyrosinemia types 1 and 2 will be added. These are important diseases with completely different prognoses if they are screened and detected in time, and their treatments initiated in time, in addition to some specific treatments such as in the case of tyrosinemia type 1, are essential for the proper clinical and biochemical control of patients.
Keywords: metabolic disorders, expanded newborn ccreening, maple syrup urine disease, homocystinuria, tyrosinemia, nitisinone
Introducción
Las ampliaciones realizadas en el cribado neonatal en el último año y las que están programadas para los próximos meses en Canarias son, y van a ser, muy importantes. Hasta finales de 2021, recordemos, se realizaba cribado de 5 metabolopatías de pequeña molécula, sin contar hipotiroidismo congénito (HTC), anemia de células falciformes (ACF) y Fibrosis quística (FQ): fenilcetonuria (PKU), aciduria glutárica tipo 1 (AG-1), déficit de acil-CoA deshidrogenasa de cadena media (MCAD), déficit de 3-OH-acilCoA deshidrogenasa (LCHAD) y deficiencia de biotinidasa (DB). En 2022 se han incorporado otras dos enfermedades: enfermedad de orina con olor a jarabe de arce (MSUD, en sus siglas en inglés) y homocistinuria (HCY) y para el año actual, en su primer semestre, serán incorporadas el cribado de la tirosinemia, tanto tipo 1 como tipo 2, ésta última por su importancia en las islas, como veremos más adelante.
Tal y como se realizó en el anterior artículo de este grupo de trabajo1, cada enfermedad será desglosada en Fisiopatología, formas de diagnóstico, tanto en cribado como clínico, tratamientos actualmente disponibles, tanto en fase de descompensación como en fase de mantenimiento, así como pronóstico y evolución.
Enfermedad de orina con olor a jarabe de arce o leucinosis (MSUD)
La leucina, isoleucina y valina son aminoácidos de cadena ramificada (branched chain amino acids, BCAA) y constituyen cerca del 40 % de los aminoácidos esenciales en el ser humano. En contraste con otros aminoácidos, los BCAA no se metabolizan en el intestino o hígado, sino en los tejidos periféricos como músculo esquelético, corazón, tejido adiposo y riñón, y por ello, su concentración en plasma aumenta de manera considerable después de comer. Su principal ruta metabólica es su incorporación a proteínas corporales (cerca de un 80%) y el 20% restante se degrada irreversiblemente por la ruta catabólica en la que comparten los dos primeros pasos, una transaminación reversible catalizada por la BCAA aminotransferasa que da lugar a sus correspondientes cetoácidos de cadena ramificada; y un segundo paso de decarboxilación oxidativa irreversible dependiente de pirofasfato de tiamina, por el complejo multienzimático mitocondrial, deshidrogenasa de los cetoácidos de cadena ramificada (branched chain keto acid dehydrogenase, BCKAD). A partir de aquí las vías metabólicas de los BCAA divergen. La enfermedad de orina con olor a jarabe de arce (MSUD, OMIM #248600) es una aminoacidopatía producida por la deficiencia del complejo multienzimático BCKAD, resultando en un acúmulo de los cetoácidos correspondientes α-ceto-isocaproico, α-ceto-β-metilvalérico y α-ceto-isovalérico y de sus BCAA precursores. Su incidencia es de 1/185.000 recién nacidos y se hereda con carácter autosómico recesivo2.
En función de la presentación clínica, la tolerancia a proteínas de la dieta, la respuesta a la administración de tiamina y la actividad enzimática residual, la MSUD se clasifica en cinco fenotipos clínicos diferentes:
- Comienzo neonatal o clásica es la más frecuente (75 % de los pacientes) y se caracteriza por un período asintomático tras el nacimiento, que puede durar una o dos semanas, dependiendo del grado de deficiencia enzimática y no necesariamente de la cantidad de proteínas ingeridas, tras el cual se inicia un cuadro clínico caracterizado por succión débil, rechazo de la alimentación y letargia que van progresivamente intensificándose; aparece el característico olor dulzón en orina, bradicardia y bradipnea; puede haber hipotonía troncular con hipertonía de extremidades, movimientos de boxeo o pedaleo y postura en opistótonos. El cuadro clínico progresa a coma y muerte si no se inicia precozmente el tratamiento. Estos síntomas neurológicos, junto con la cetosis (raramente con acidosis), con ácido láctico y amonio normales constituyen la forma más frecuente de presentación. La actividad enzimática residual es del 0-2% con respecto a la normal.
- Forma intermedia, muy rara, puede iniciarse desde los 5-6 meses hasta los 6-7 años de edad, con síntomas neurológicos progresivos: retraso psicomotor, convulsiones y ataxia; la actividad enzimática residual es del 3-30 %.
- Forma intermitente puede aparecer a cualquier edad, caracterizada por síntomas neurológicos tipo intoxicación semejantes a la forma clásica, desencadenados por situaciones de catabolismo (infecciones, quemaduras, intervenciones quirúrgicas) y que pueden ser letales, siendo la actividad residual enzimática entre el 5 y 20 %.
- Formas sensibles a tiamina, aunque no existe un criterio uniforme para el diagnóstico de esta rara forma, tiene una presentación similar a la forma intermitente.
- Deficiencia de dihidrolipoil deshidrogenasa (componente E3), que es uno de los componentes del complejo multienzimático BCKAD, tiene una clínica similar a la forma intermedia; la actividad residual enzimática oscila entre el 0 y 25 %3.
El diagnóstico se realiza con la determinación plasmática de los aminoácidos ramificados. En la forma clásica la leucina puede ser superior a 2.000 µmol/L (normal 80-200 µmol/L), y es característica la presencia de aloisoleucina, compuesto resultante de la racemización no enzimática de la isoleucina. En orina aparece un aumento de los ácidos 2-OH-isocaproico, 2-OH-3-metilvalérico y 2-OH-isovalérico. Mediante cribado neonatal se realiza con el aumento de los BCAA y del cociente leucina/isoleucina en sangre seca, por espectrometría de masas en tándem.
La demostración del defecto enzimático se realiza de manera indirecta midiendo la descarboxilación de [1-14C] leucina en fibroblastos cultivados. En el análisis del defecto genético pueden existir mutaciones en los genes BCKDHA, BCKDHB, DBT, DLD, PPM1K Y BCAT24.
El objetivo del tratamiento en la fase aguda es la rápida normalización de los niveles de BCAA, en especial de la leucina que es el más neurotóxico, y posteriormente un tratamiento dietético de mantenimiento que consiga un adecuado crecimiento y desarrollo del paciente, al mismo tiempo que evite un aumento significativo de los niveles de BCAA. Así pues, en la fase aguda, el tratamiento debe ser inmediato debido al peligro de daño neurológico irreversible e incluso muerte. Existen dos alternativas para reducir lo más rápidamente posible las altas concentraciones de BCAA, que tienen un bajo aclaramiento renal:
- Técnicas extracorpóreas: la de elección es la hemodiálisis con hemofiltración y sólo excepcionalmente diálisis peritoneal o exanguinotransfusión. Estas técnicas consiguen unos descensos significativos, rápidos y mantenidos de los BCAA, pero requieren una infraestructura compleja y el acceso vascular es difícil en neonatos y lactantes. La elección de la técnica a emplear debe depender de la experiencia y disponibilidades de cada hospital.
- Incorporar el exceso de BCAA a la síntesis proteica mediante la inducción al anabolismo con un tratamiento nutricional agresivo con alimentación parenteral y/o nutrición enteral continua. La nutrición parenteral debe realizarse con una mezcla de aminoácidos exenta de BCAA (no disponible en España) y un alto contenido energético con glucosa e insulina si fuera necesario, así como lípidos. Otra alternativa eficaz es iniciar una nutrición enteral a débito continuo con una fórmula exenta de BCAA intentando conseguir un aporte proteico entre 2.5-3 g/kg/día (según la edad), y por lo menos, los requerimientos energéticos normales también según la edad. Esto a veces es difícil de alcanzar en los primeros días, por lo que es necesario empezar con una fórmula diluida y suplementada con polímeros de glucosa hasta una concentración final de un 10% de carbohidratos, o bien combinar la nutrición enteral con glucosa intravenosa junto con insulina (si fuera necesaria), para conseguir un aporte energético alto y evitar el catabolismo.
- La combinación de ambas técnicas (técnicas extracorpóreas junto a soporte nutricional) disminuye más rápidamente los niveles plasmáticos de leucina en un plazo de 12-24 horas.
- Tratamiento del edema cerebral: osmolaridad, manitol, furosemida, ondansetrón (vómitos).
- Tratamiento específico de la causa desencadenante5
En la fase de mantenimiento, el objetivo es conseguir un adecuado crecimiento y desarrollo previniendo las deficiencias de macro y micronutrientes, y evitar y tratar las crisis de descompensación. Los aportes de BCAA en cada paciente deben basarse en sus concentraciones plasmáticas, que deben mantenerse, cuando el paciente está asintomático en: leucina 100-200 µmol/l (1.3-2.6 mg/dl), isoleucina 200-400 µmol/l, y valina 200-400 µmol/l.
Las necesidades de leucina oscilan entre 50-90 mg/kg/día en los 2-3 primeros meses de vida, disminuyendo a lo largo del primer año hasta 40-50 mg/kg/día. La mayoría de los niños afectos de MSUD tienen una ingesta de leucina entre 400-600 mg/día. Con frecuencia, al mantener las concentraciones de leucina en el rango recomendado, las de isoleucina y valina descienden a < 100 µmol/l, y es necesario suplementarlos ambos a dosis de 100-200 mg/kg/día para evitar signos clínicos de carencia proteica.
Cuando el diagnóstico es mediante cribado neonatal los objetivos se establecen basándose en los niveles de leucina al diagnóstico y la clínica inicial del paciente. La restricción de proteínas naturales de la dieta obliga al empleo de suplementos proteicos libres de BCAA para completar el aporte proteico necesario para un adecuado crecimiento y desarrollo cognitivo en combinación con la lactancia materna o fórmula infantil. La mayoría de estos preparados contienen vitaminas y minerales que cubren las necesidades del paciente según la edad. Cuando se inicia la diversificación alimentaria se emplean alimentos de bajo contenido proteico, y para alcanzar las necesidades calóricas se emplean aceites y módulos de dextrinomaltosa y/o lípidos. En el seguimiento de los pacientes, se realiza con la monitorización clínica del crecimiento, bioquímica con los niveles de aminoácidos plasmáticos y dietética6.
Se recomienda un suplemento de tiamina de 5 mg/kg/día para todas las formas de la enfermedad, puesto que pueden mejorar la tolerancia a los BCAA en algunos pacientes. Las formas sensibles a tiamina requieren entre 10-1.000 mg/día7.
En las crisis de descompensación metabólica, durante las infecciones intercurrentes o los procesos de mayores demandas, se produce un rápido aumento de los BCAA, sobre todo de la leucina, sin que existan alteraciones en los estudios bioquímicos rutinarios (glucemia, lactato, amonio, pH). Por ello, es necesario observar la posible presencia de síntomas inespecíficos como hipotonía/hipertonía, decaimiento alternando con agitación, somnolencia, alteraciones en el lenguaje, etc. En estos casos, es necesario suprimir la ingesta de alimentos proteicos e iniciar un régimen de emergencia con polímeros de glucosa junto con el preparado proteico exento de BCAA para promover la síntesis proteica y proporcionar un adecuado aporte calórico, y valorar la derivación a un centro hospitalario8.
Homocistinuria
El resumen del paciente con homocistinuria clásica podría ser el de una persona con fenotipo Marfan y fenómenos tromboembólicos, además de retraso mental de diversa gravedad. Esta enfermedad, consecuencia de la alteración del metabolismo de la metionina (en su vía metabólica de trans-sulfuración y remetilación), de herencia autosómica recesiva, se pueden ver afectados los genes CBS, MTHFR, y los genes de la cobalamina C, D y F que asocian aciduria metilmalónica. Entró, tal y como se ha comentado al inicio, en nuestro programa de cribado a finales de 2022, con la determinación de la Metionina (Met) ya que, al contrario que las otras posibles causas, la Met está elevada en el caso de la homocistinuria clásica (CBS, 21q22.3, OMIM #613381)9, y ese es el objetivo del cribado. Descrita en 1962 por Carson y Neill10, se ha descrito en España, Portugal y países de Sudamérica, un predomino de la mutación T191M, al contrario que en otros países europeos, asociada a una no respuesta al tratamiento con piridoxina (B6)11. En la actualidad, se conoce que el aumento de la homocisteína en plasma es la causante de las manifestaciones clínicas: fallo de medro, retraso psicomotor, alteraciones vasculares, oculares y complicaciones tromboembólicas (accidentes cerebro-vasculares), principales causas de morbi-mortalidad de esta enfermedad12. Hay que destacar que dentro de las alteraciones oculares con luxación/ectopia del cristalino, para diferenciarlas de las producidas en el Síndrome de Marfan, la luxación inferior es debida generalmente por la homocistinuria y la superior al Marfan.
Entre las causas de origen adquirido, cabe destacar, como causa más frecuente en la infancia, la combinación de hiperhomocisteinemia con aciduria metilmalónica en los hijos de madres con LM exclusiva y que éstas tengan un déficit de factor intrínseco no tratadas o una dieta vegetariana estricta.
Los pacientes con déficit de CBS presentan retraso mental en hasta un 50% de los casos (otra diferencia con el síndrome de Marfan), pero que no llega a ser grave es muy raro que lo sea, y cuyo origen está en una disminución de la cistationina, inhibición competitiva con otros AA y el problema de la formación de neurotransmisores, entre otros. Las convulsiones pueden estar presentes en un 20%, así como alteraciones del comportamiento o síntomas esquizoides.
En cuanto al sistema esquelético, son frecuentes la osteoporosis, escoliosis, fracturas patológicas, colapso vertebral, pero que se diferencian del Marfan, nuevamente, en que estos pacientes sí tienen limitación articular. Los pacientes con Marfan presentan más hiperlaxitud articular13.
El diagnóstico actual se inicia en el cribado neonatal, con la determinación de Metionina y ratio Met/Phe, pero tiene el inconveniente de presentar una alta tasa de falsos negativos, para lo cual se incluye la homocisteína como determinación de 2º nivel o bajar el dintel para considerar positivo de la metionina. En caso de positividad, como pruebas confirmatorias se realiza la homocisteína, tanto en plasma como en orina, y Met (mediante analizador de AA, no mediante MS/MS). Posteriormente, ante la positividad de las pruebas realizadas, podemos pasar a análisis enzimático de la CBS o directamente a la genética9.
El tratamiento de la homocistinuria no es una terapia específica, aunque actualmente hay en proceso un estudio clínico con una variante humana enzimática de la CBS-pegilada (OT-58, https://clinicaltrials.gov/ct2/show/NCT03406611). El tratamiento consiste en aportar tanto piridoxina (B6, cofactor de la CBS)14, ácido fólico y ácido folínico, cobalamina (B12) y betaína (trimetilglicina)15, que actúa como cofactor en la remetilación. La dieta, como es obvio, debe ser casi exenta en Met, aunque puede recibir lactancia materna de forma controlada combinada con aportes selectivos de AA y suplementos de L-cisteína, manteniendo el resto de macro y micronutrientes según las recomendaciones internacionales16. Otros tratamientos como aspirina y dipiramol no han demostrado eficacia alguna, la vitamina C puede mejorar la disfunción endotelial y en estos pacientes debemos evitar actividades que aumenten el riesgo tromboembólico, como ACO, cirugías o la deshidratación7.
El seguimiento de estos pacientes debe incluir controles cada 1-3 meses de Hcy (< 40-50 μmol/L), anuales de oftalmología y cardiología, B12 y ácido fólico, densitometría y marcadores de osteopenia. El pronóstico, sin tratamiento, hasta el 25% fallecen antes de los 30 años. Los respondedores a B6 mejoran síntomas, los heterocigotos tienen mayor probabilidad de enfermedad oclusiva vascular y cerebral prematura.
La vacuna para la COVID-19, que en general, y según el documento de consenso de los profesionales clínicos de la Asociación Española para el Estudio de los Errores Congénitos del Metabolismo (AECOM), es de destacar que para todas las enfermedades metabólicas y los pacientes “…no deben considerarse como “de alto riesgo” frente a este tipo de vacuna. Excepciones pueden ser los pacientes con riesgo de trombosis o microangiopatía trombótica como la homocistinuria, defectos de la cobalamina o del folato, o pacientes con grave disfunción endotelial o plaquetaria (pacientes con enfermedad mitocondrial o acidemia orgánica descompensadas, enfermos lisosomales con grave secuestro esplénico, defectos de glicosilación o algunas glucogenosis Ia o Ib)” (Documento de consenso de los profesionales clínicos de AECOM, 18 de marzo de 2021: https://aecom.com.es/wp-content/uploads/2021/03/Posicionamiento-AECOM.pdf), con lo que estos pacientes deben extremarse las precauciones y realizar de manera individualizada la indicación para recibir dicha vacuna.
Tirosinemia tipos 1 y 2
La tirosinemía tipo 1 (TYRSN1) o tirosinemia hepato-renal es un EIM del metabolismo de la tirosina (Tyr), AA no esencial que se obtiene de los alimentos y del AA Phe, mediante la enzima fenilalanina hidroxilasa (PAH), ruta metabólica ya vista en artículos anteriores1. De herencia autosómica recesiva, su incidencia se estima en 1:100.000 RN. La tirosina puede seguir tres vías:
- Precursora de la síntesis de catecolaminas, melanina y hormonas tiroideas.
- Otra de degradación hacia fumarato (ciclo de Krebs y gluconeogénesis) y acetoacetato (síntesis lipídica y obtención de acetil-CoA).
- Formación de proteínas. Es un aminoácido importante que, si se ve comprometido en alguna parte de su metabolismo, puede dar lugar no sólo a las tirosinemias, sino también a la alcaptonuria, albinismo o enfermedad de Parkinson, entre otras.
En el caso de la tirosinemia tipo 1, el gen defectuoso es la fumarilacetoacetato hidrolasa (FAH, 15q25.1, OMIM #276700), en la vía de degradación de la Tyr, en su paso de fumarilacetoacetato a fumarato y acetato (último paso del proceso de degradación). Al producirse este bloqueo, el producto acumulado (fumarilacetoacetato) toma la vía alternativa hacia succinilacetoacetato (SAA) y succinilacetona (SA), siendo ésta última un inhibidor competitivo de la enzima ácido δ-aminolevulínico dehidratasa (ALAD, en sus siglas en inglés). Esta inhibición resulta en la acumulación del ácido δ-aminolevulínico (ALA) y su excreción en la orina. El ALA se ha asociado con toxicidad mitocondrial, toxicidad hepática, cáncer de hígado y problemas neuro-psiquiátricos. A nivel renal, la SA causa una disfunción Fanconi-like por alteración de la membrana renal y disfunción tubular17.
La presentación clínica de la tirosinemia tipo 1 es variable y depende de la actividad enzimática. A menor actividad, mayor gravedad. En los casos graves, se puede producir a las pocas semanas fallo hepático agudo con aparición de vómitos, sangrado, hipoglucemia, alteraciones de la coagulación, ascitis, ictericia y hepatomegalia, que puede conllevar una alta mortalidad. En el caso de las alteraciones de la coagulación, en niños sin datos de fallo hepático, se debería sospechar tirosinemia tipo 118, al igual que la cirrosis o la aparición de hepatocarcinoma en la etapa infantil, ya que es la principal causa en edades tempranas, con picos de incidencias entre los 4-5 años19. La disfunción renal, ya comentado el síndrome Fanconi-like, se caracteriza por glucosuria y raquitismo hipofosfatémico, que en última instancia conlleva a la nefrocalcinosis20.
Dentro de los parámetros bioquímicos habituales que se pueden detectar en los pacientes previo al diagnóstico etiológico son los de fallo hepático con datos de coagulopatía con TTPa y TP elevados, hipoalbuminemia, hipertransaminasemia, hiperbilirrubinemia, hiperamoniemia que puede ser moderada, AFP muy elevada; afectación tubular e hipoglucemia. La Tyr se puede encontrar elevada, aunque no es el marcador específico ya que puede elevarse en niños prematuros, nutrición parenteral total, síndromes de depleción mitocondrial u otras enfermedades metabólicas como las tirosinemias tipo II y III, como una hipertirosinemia transitoria del recién nacido; aumento de metionina y excreción aumentada del ácido δ-aminolevulínico en orina y SA tanto en sangre como en orina, siendo ésta última la clave para el diagnóstico y marcador utilizado en el cribado neonatal. El estudio genético sobre el gen FAH confirma el diagnóstico21,22. En España la mutación más frecuente el cambio intrónico IVS6-1(G > T) presente en hasta el 66.6% de los pacientes en el último estudio realizado23.
Además de la restricción proteica, con uso de fórmulas de aminoácidos exentas en Phe y Tyr, manteniendo el resto de macro y micronutrientes según las recomendaciones internacionales16, el pronóstico de los pacientes con tirosinemia tipo 1 ha cambiado radicalmente tras la introducción de la nitisinona (NTBC, herbicida)24, sustancia que realiza un bloqueo del paso previo de la FAH, permitiendo un acúmulo del 4-OH-fenilpirúvico, menos dañino, pero que secundariamente aumenta la Tyr, pudiendo ocasionar cristalización de la misma en la córnea y toxicidad a nivel cerebral. Para evitar esto, se deben mantener unos Niveles óptimos de Tyr en plasma entre 200 y 400 μmol/L. La dosis recomendada es de 1-2 mg/Kg/día, administrándose cada 12-24 horas. Se dispone de cápsulas de 5, 10 y 20 mg, y jarabe mg/mL. La dieta es restrictiva en proteínas, con unas necesidades de fenilalanina y de tirosina entre 30-100 mg/Kg/día, siendo muy parecida a la fenilcetonuria, pero algo más laxa. El trasplante hepático que puede ser muy urgente con código 0 si no hay respuesta a NTBC y se produce un fallo hepático agudo, es la última alternativa, quedando posteriormente el paciente con una inmunodepresión para evitar rechazo durante toda su vida21.
El pronóstico suele ser bueno, con una buena calidad de vida, una sola medicación y dieta de por vida, con buena integración escolar, pudiendo aparecer en la adolescencia problemas de hiperactividad y neurocognitivos.
La tirosinemia tipo 2 (TYRSN 2, OMIM #276600) tiene una importancia relevante en Canarias. En la isla de Gran Canaria se han detectado hasta 5 pacientes de familias distintas con una mutación predominante (p.P406L) tipo “efecto fundador” (presente en los pacientes en homocigosis) no descrita hasta el momento de la publicación25. También conocida como síndrome Richner-Hanhart, presenta una muy alta incidencia como se ha mencionado, en la isla de Gran Canaria, ya que normalmente suele ser de 1:250.000 RN. Se afecta el gen de la tirosina aminotransferasa (TAT, 16q22.2), primer paso en la degradación de la Tyr, y cuyo acúmulo tóxico importante es la propia Tyr ya que, como se mencionó anteriormente en el tratamiento con NTBC en la tipo 1, la Tyr forma cristales en córnea (queratitis bilateral, herpes-like) y epidermis (lesiones hiperqueratósicas, callos). Además, puede haber afectación neurológica leve-moderada a largo plazo.
El diagnóstico sin cribado neonatal se realiza de forma clínica por los hallazgos encontrados y posteriormente con la medición de la Tyr en sangre. Mediante cribado, las cifras elevadas de Tyr al nacimiento son las que ponen sobre aviso para su estudio. Recordemos que hay otras causas de aumento de Tyr en sangre que hay que descartar mediante segundas muestras y marcadores secundarios (Phe/Tyr disminuido). En ambos casos, la SA será normal EL estudio molecular EN EL gen TAT confirmará el diagnóstico26.
El tratamiento de la TYRSN2 está limitado a la restricción proteica, manteniendo cifras de Tyr en sangre entre 200-500 μmol/L, además de fórmulas especiales con aminoácidos y restringidas en Phe y Tyr, manteniendo el resto de macro y micronutrientes según las recomendaciones internacionales16. El simple hecho de bajar las cifras de Tyr en sangre revierte los efectos de la misma a nivel ocular y cutánea, pudiendo, si se mantiene estable, vivir sin mayores complicaciones27. Se han comunicado casos de embarazos que con niveles de Tyr > 1000 μmol/L, pueden afectar al feto produciendo microcefalia, convulsiones y retraso mental. Si esos niveles estaban por debajo de 200, no afectaban y se han producido casos con niños normales26.
En Canarias su introducción en cribado es obligatoria por la alta incidencia a la zona de Gran Canaria, incluso mayor que la tipo 1, evitando así las manifestaciones clínicas, diagnóstico precoz y consejo genético adecuado25.
Conclusiones
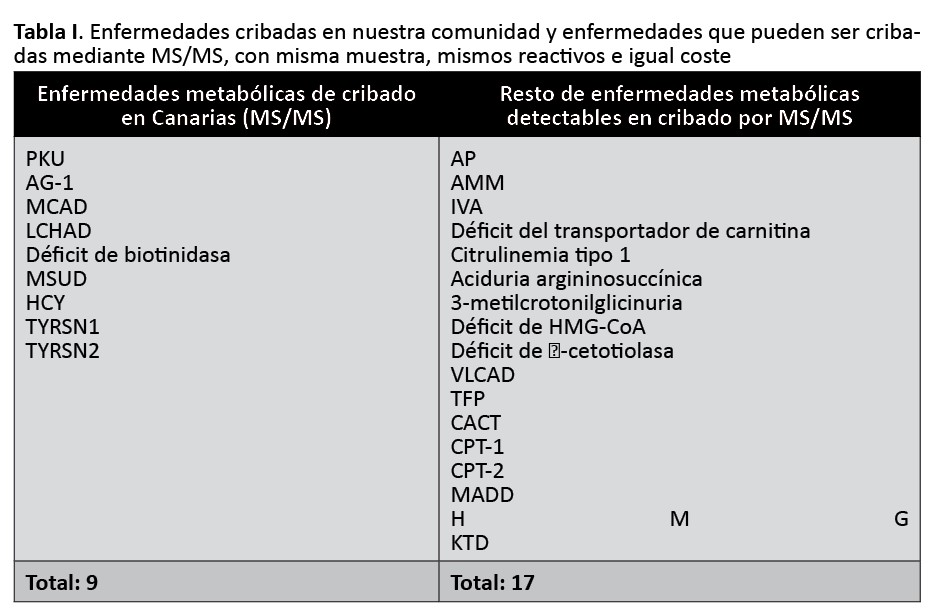
El CNA en Canarias avanza hacia una mejora necesaria y harto solicitada. Con la adición de estas cuatro nuevas enfermedades, se sitúa en un total de nueve para metabolopatías específicas, acorde a los requisitos mínimos determinados por el consejo interterritorial en su última reunión en abril de 2022 y que modifican el RD 1030/2006 de la cartera de servicios del SNS. No obstante, estos avances siguen siendo escasos en nuestro medio. La tecnología de espectrometría de masas en tándem, nos permite cribar la mayoría de aminoacidopatías y defectos de la beta-oxidación con la misma muestra y reactivos, sin producirse un aumento en el coste al incluir todas las enfermedades restantes28. Para hacerse una idea, la tabla I define las enfermedades cribadas y las que se podrían cribar, repitiendo, por el mismo coste.
Seguir las directrices del consejo interterritorial, y al ritmo actual de 2 enfermedades/año nos abocaría a mínimo 10-15 años de retraso para tener un cribado completo. Este grupo de trabajo considera que mantener este proceso es contraproducente y carente de todo sentido, pues:
- En la mayoría de comunidades (13 de 20 laboratorios de toda España) donde se ha implementado el cribado ampliado por MS/MS han comenzado con un mayor número y no limitadas a lo indicado por el Ministerio de Sanidad
- Esta disparidad se hace patente en que podemos encontrar laboratorios con siete enfermedades y otros con más de 20, comprometiendo seriamente la igualdad entre los ciudadanos de este país29
- Basar la implantación de enfermedades en el cribado en informes de coste-efectividad realizados por la Red Española de Agencias de Evaluación de Tecnologías Sanitarias (AETS, https://redets.sanidad.gob.es/) en el caso de enfermedades raras cribadas mediante MS/MS, donde añadir no implica una modificación del coste del cribado, supone abocar a dichos pacientes a un retraso del diagnóstico, tratamiento y del consejo genético, sin poder evitar, en determinados casos, nuevos nacimientos con la enfermedad heredada
- Debido a este retraso del diagnóstico, agravado en nuestra comunidad por su situación geográfica, hace que los diagnósticos de EIM en Canarias sean, si cabe, más complicados y tardíos, con lo que ello conlleva en la situación clínica del paciente, confianza y seguridad de su familia en el sistema.
En el caso de los informes de cribado mediante MS/MS realizados por las AETS en los que se basa el Ministerio de Sanidad para la inclusión de las enfermedades en el cribado, si bien su realización en este tipo de enfermedades puede ser discutible, hay retrasos palpables y duplicación del trabajo que hacen que la inclusión de nuevas enfermedades se vea comprometida. Como ejemplo, trabajos realizados en 2013 y 2014 sobre MSUD, HCY, AG-1, LCHAD, tirosinemia tipo 1 y acidemias orgánicas, siendo algunas incluidas hasta 8 años después, y realizándose un nuevo informe en 2020 por parte del equipo de trabajo de Galicia repitiendo el análisis sobre MSUD, isovalérica y HCY e inicio de “estudio piloto”. Galicia lleva desde el año 2000 cribando estas enfermedades, con la mayor incidencia de MSUD (1/39.300 RN) de España30. Si Galicia no hubiese cribado desde el año 2000, ¿Qué hubiese pasado con esos pacientes?
Haciendo mención en el resumen del informe del 2020: “la inclusión de una enfermedad en un programa de cribado debe realizarse de manera racional y eficiente. Los programas de cribado deben disponer de suficiente evidencia científica sobre los beneficios en salud y de estudios de coste-efectividad”. Duane Alexander director del National Institute of Child Health and Human Development (NICHD, EEUU), fallecido en el 2020 por enfermedad de Alzhehimer dijo “Criba, a no ser que haya una apremiante razón para no cribar” y “centrarse en lo que no hay que cribar, más que en lo que hay que incluir”31. Este grupo de trabajo es partidario de esto último y recomienda e insiste en la inclusión completa de los EIM detectables en MS/MS, acorde a lo realizado en otras comunidades, de la forma más rápida que técnica y humanamente sea posible. No quedan muchas y es obvio que el camino seguido por el Ministerio de Sanidad llevará a la misma conclusión, pero para posiblemente “sólo algunos” pacientes podría ser demasiado tarde en nuestra comunidad. Por muy pocos que sean, siempre merece la pena realizarlo.
Además, queremos transmitir la imperiosa necesidad de que el actual laboratorio de cribado neonatal se convierta en el laboratorio de referencia para el estudio diagnóstico de sospecha de EIM. Para ello es necesario una hoja de ruta clara y comenzar a analizar, no sólo las muestras de cribado, sino de todos aquellos niños con metabolopatías diagnosticadas (actualmente sólo se hace en el control de Phe en niños con fenilcetonuria) para control metabólico y de aquellas sospechas que surjan en los hospitales de referencia de nuestra comunidad. Así mismo, es muy importante la formación de todos los profesionales sanitarios implicados en este proceso. Una recogida de muestra correcta, con envío adecuado en tiempo y forma, y conocer las repercusiones que tiene en el cribado de este tipo de enfermedades, es de imperiosa necesidad de conocimiento para todos los implicados y para el correcto funcionamiento del laboratorio y el cribado que realiza.
Esta es la única solución real para la increíble problemática actual de tiempos sospecha/extracción/transporte/análisis/resultados/inicio de tratamiento específico que tenemos y que, a día de hoy, siendo de extrema importancia y enorme magnitud para los profesionales, pacientes y familias, sigue sin solución.
Bibliografía
- Valerio Hernández E, González Delgado A, Rodríguez Fernández-Oliva CR, Murray Hurtado M, Peña Quintana L, Ruiz Pons M. Actualización del cribado neonatal ampliado en Canarias. Nuevas enfermedades y perspectivas futuras. Can Pediatr 2022, 46:92-104. Disponible en: https://scptfe.com/actualizacion-del-cribado-neonatal-ampliado-en-canarias-nuevas-enfermedades-y-perspectivas-futuras/
- Blackburn PR, Gass JM, Vairo FP E, Farnham KM, Atwal HK, Macklin S et al. Maple syrup urine disease: mechanisms and management. Appl Clin Genet 2017; 10:57. Disponible en: /pmc/articles/PMC5593394/
- Strauss KA, Carson VJ, Soltys K, Young ME, Bowser LE, Puffenberger EG et al. Branched-chain α-ketoacid dehydrogenase deficiency (maple syrup urine disease): Treatment, biomarkers, and outcomes. Mol Genet Metab 2020; 129:193-206. Disponible en: https://doi.org/10.1016/j.ymgme.2020.01.006
- Strauss KA, Puffenberger EG, Carson VJ. Maple Syrup Urine Disease. En: Gene Rev. Pagon R, Adam M, Ardinger H, eds. 2020. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK1319/
- Vitoria I, Merinero B, Sánchez-Valverde F, Gil D, Dalmau J. Enfermedad de orina de jarabe de arce. En: Protocolos de diagnóstico y tratamiento de los errores congénitos del metabolismo. Madrid: Ergon 2018. pp. 85-94. Disponible en: https://aecom.com.es/wp-content/uploads/2018/01/protocolos-AECOM-2-ed.pdf
- Ruiz Pons M, Sanchez Valverde F, Dalmau Serra J. Tratamiento nutricional de los errores innatos del metabolismo. Madrid: Ergon 2015. Disponible en: https://ergon.es/wp-content/uploads/2015/08/n018_Tratam_nutricional_EIM_3ed.pdf
- Valerio Hernández E, Ruíz Pons M, Peña Quintana L, Murray Hurtado M, Rodríguez Fernández-Oliva CR, de la Nuez Viera F, Plasencia Núñez M. Errores congénitos del metabolismo herramientas terapéuticas. Boletín Canar Uso Racion del Medicam del SCS 2021; 13:1-7. Disponible en: https://www3.gobiernodecanarias.org/sanidad/scs/content/f4657c2e-4160-11ec-bda3-d3e6cfb1ac5c/BOLCAN ECM V13_n2_set2021.pdf
- Alcalde Martín C, de las Heras Montero J, Llarena Fernández M, Andrade Lodeiro F. Enfermedad de jarabe de arce (Maple Syrup Urine Disease, MSUD). En: Enfermedades raras metabólicas Procedimientos de urgencias y de situaciones de riesgo. Madrid: Ergon 2017, pp. 211-219. Disponible en: https://aecom.com.es/wp-content/uploads/2020/03/PROTOCOLO-DE-URGENCIAS.pdf
- Morris AAM, Kožich V, Santra S, Andria G, Ben-Omran TIM, Chakrapani AB et al. Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency. J Inherit Metab Dis 2016; 40:49-74. Disponible en: https://link.springer.com/article/10.1007/s10545-016-9979-0
- Carson NAJ, Neill DW. Metabolic abnormalities detected in a survey of mentally backward Individuals in Northern Ireland. Arch Dis Child 1962; 37:505. Disponible en: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2012909/
- Urreizti R, Asteggiano C, Bermudez M, Córdoba A, Szlago M, Grosso C et al. The p.T191M mutation of the CBS gene is highly prevalent among homocystinuric patients from Spain, Portugal and South America. J Hum Genet 2006; 51:305-313
- García-Jiménez MC, Baldellou A, García-Silva MT, Dalmau-Serra J, García-Cazorla A, Gómez-López L et al. Estudio epidemiológico de las enfermedades metabólicas con homocistinuria en España. An Pediatr 2012; 76:133-139
- Couce Pico ML, González Vioque E, Dionisi-Vici C. Trastorno del metabolismo de los aminoácidos azufrados. En: Couce Pico ML, Aldámiz-Echevarría L, García Jiménez MC, González-Lamuño Leguina D, eds. Diagnóstico y Tratamiento de las enfermedades metabólicas hereditarias, 5a Madrid: Eegpn 2022, pp. 367-376
- Kožich V, Sokolová J, Morris AAM, Pavlíková M, Gleich F, Kölker S et al. Cystathionine ß-synthase deficiency in the E-HOD registry-part I: pyridoxine responsiveness as a determinant of biochemical and clinical phenotype at diagnosis. J Inherit Metab Dis 2021; 44:677-692
- Valayannopoulos V, Schiff M, Guffon N, Nadjar Y, García-Cazorla A, Martinez-Pardo Casanova M et al. Betaine anhydrous in homocystinuria: Results from the RoCH registry. Orphanet J Rare Dis 2019; 14:1-10
- Hermoso M, Tabacchi G, Iglesia-Altaba I, Bel-Serrat S, Moreno-Aznar LA, García-Santos Y et al. The nutritional requirements of infants. Towards EU alignment of reference values: The EURRECA network. Matern Child Nutr 2010; 6:55-83
- Morrow G, Tanguay RM. Biochemical and clinical aspects of hereditary tyrosinemia Type 1. Adv Exp Med Biol 2017; 959:9-21. Disponible en: https://pubmed.ncbi.nlm.nih.gov/28755181/
- JM C, SK G, SK C, JF F. Tyrosinemia type 1 should be suspected in infants with severe coagulopathy even in the absence of other signs of liver failure. Pediatrics 1999; 103:675-678. Disponible en: https://pubmed.ncbi.nlm.nih.gov/10049978/
- van Ginkel WG, Pennings JP, van Spronsen FJ. Liver cancer in tyrosinemia type 1. Adv Exp Med Biol 2017; 959:101-109. Disponible en: https://pubmed.ncbi.nlm.nih.gov/28755188/
- Brito dos Santos S, Bertholet-Thomas A, Butin M, Dubourg L, Fouilhoux A, Bacchetta J. Tyrosinemia type 1 in pediatric nephrology: Not always straightforward. Arch Pediatr 2021; 28:338-341. Disponible en: https://pubmed.ncbi.nlm.nih.gov/33858731/
- de Laet C, Dionisi-Vici C, Leonard J V., McKiernan P, Mitchell G, Monti L et al. Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis 2013; 8:8. Disponible en: https://pubmed.ncbi.nlm.nih.gov/23311542/
- Chinsky JM, Singh R, Ficicioglu C, van Karnebeek CDM, Grompe M, Mitchell G et al. Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations. Genet Med 2017; 19:1380-1395
- Couce ML, Dalmau J, del Toro M, Pintos-Morell G, Aldámiz-Echevarría L. Tyrosinemia type 1 in Spain: mutational analysis, treatment and long-term outcome. Pediatr Int 2011; 53:985-989. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/21752152
- Couce ML, Sánchez-Pintos P, Aldámiz-Echevarría L, Vitoria I, Navas V, Martín-Hernández E et al. Evolution of tyrosinemia type 1 disease in patients treated with nitisinone in Spain. Medicine (Baltimore) 2019; 98: e17303. Disponible en: https://pubmed.ncbi.nlm.nih.gov/31574857/
- Peña-Quintana L, Scherer G, Curbelo-Estévez ML, Jiménez-Acosta F, Hartmann B, La Roche F et al. Tyrosinemia type II: Mutation update, 11 novel mutations and description of 5 independent subjects with a novel founder mutation. Clin Genet 2017; 92:306-317. Disponible en: https://onlinelibrary.wiley.com/doi/full/10.1111/cge.13003
- Couce Pico ML, Aldámiz-Echevarría Azuara L. Trastornos del metabolismo de la Tirosina. Tirosinemia tipo I, II, III, hawkinsinuria, alcaptonúrias y deficiencia de la maleilacetoacetato isomerasa. En: Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias, 5a. Couce ML, Aldámiz-Echevarría L, García-Jiménez MC, González-Lamuño D, eds. Madrid: Eegon 2022, pp. 357-365
- Nakamura K, Matsumoto S, Mitsubuchi H, Endo F. Diagnosis and treatment of hereditary tyrosinemia in Japan. Pediatr Int 2015; 57:37-40. Disponible en: https://pubmed.ncbi.nlm.nih.gov/25443793/
- Vitoria I, Martín-Hernández E, Peña-Quintana L, Bueno M, Quijada-Fraile P, Dalmau J et al. Carnitine-acylcarnitine translocase deficiency: experience with four cases in Spain and review of the literature. JIMD Rep 2015; 20:11-20. Disponible en: https://pubmed.ncbi.nlm.nih.gov/25614308/
- Castiñeras DE, Couce ML, Marin JL, González-Lamuño D, Rocha H. Newborn screening for metabolic disorders in Spain and worldwide. An Pediatr 2019; 91:128.e1-128.e14
- Couce Pico ML, Castiñeiras Ramos DE, Bóveda Fontán MD, Iglesias Rodríguez AJ, Cocho De Juan JA, Fraga Bermúdez JM. Advances in the diagnosis and treatment of maple syrup urine disease: experience in Galicia (Spain). An Pediatr (Barc) 2007; 67:337-343. Disponible en: https://pubmed.ncbi.nlm.nih.gov/17949643/
- Alexander D, Van Dyck PC. A Vision of the future of newborn screening. Pediatrics 2006; 117:S350-354. Disponible en: /pediatrics/article/117/Supplement_3/S350/68865/A-Vision-of-the-Future-of-Newborn-Screening